



论坛速递:小彩虹YOUYOU遗传变异配对小程序发布暨第4期遗传病案例分享解读系列圆满结束
发布日期:2023-07-28

小彩虹YOU-YOU遗传病案例分享解读系列会议第4期特别邀请了郑州大学第三附属医院高永强副主任医师担任案例分享与讨论嘉宾,复旦大学遗传咨询在读研究生万远对案例的变异进行解读,首都医科大学宣武医院神经内科王朝东主任、合因科技医学服务部总监谭灏文博士对案例深度解析和讨论,安宇老师作为主持人。
案例分享
郑州大学第三附属医院高永强副主任此次分享的是一则有关儿童遗传性神经肌肉病案例,从病史、查体、检查、诊断、治疗及随访等方面对患儿临床情况做了全面详细讲解,并综合介绍了IGHMBP2基因相关疾病。
临床表型:患儿,女,1岁3月因不能独站就诊,2岁6月时走路不稳,双下肢肌张力低,膝过伸,双足内旋,呈鸭步态,MRI显示骶管脂肪沉积。肌电图&诱发电位可见:双胫神经运动传导速度减慢,双腓总神经运动传导未引出肯定波形,双胫前肌、双腓肠肌可见纤颤电位、正锐波及巨大电位,余被检肌未见自发电位,重收缩用力可,考虑双下肢部分周围神经病变(提示脱髓鞘伴轴索损伤)。大运动能力低下。无家族史,其余方面查体结果、智力认知语言水平无异常,关节活动度未见明显异常。初步诊断:周围神经疾病,遗传性运动感觉神经病?
基因检测:为进一步明确病因,对患儿进行了家系临床全外显子组测序(Trio-CES)和靶向多重连接探针扩增技术(MLPA)检测。MLPA检测结果排除了受检者的MPZ、PMP22和GJB1基因外显子区域的缺失重复变异。二代测序发现受检者携带IGHMBP2基因上的两个杂合变异,即变异1:c.2362C>T:p.R788*和变异2:c.1910G>T:p.R637L。一代测序验证结果显示这两个变异真实可靠,且分别遗传自受检者母亲和父亲,构成复合杂合变异。根据ACMG评级,变异1为致病性变异,变异2为意义不明变异。
高永强主任对IGHMBP2基因异常相关疾病的临床特征、诊断标准、鉴别诊断以及治疗等方面进行了概述。IGHMBP2基因异常可导致常染色体隐性遗传的脊髓性远端肌萎缩1型或轴索性腓骨肌萎缩症2S型,然而患儿的临床表现并不能完全匹配此基因相关2种疾病,临床按照腓骨肌萎缩症2S型的方向进行治疗,目前孩子仍可维持独立行走的能力。
IGHMBP2基因可以导致两种不同的表型:
IGHMBP2基因异常导致常染色体隐性遗传的脊髓性远端肌萎缩 1 型((DSMA1;OMIM#604320)或轴索性腓骨肌萎缩症 2S 型(CMT2S;OMIM#616155)
DSMA1又叫脊髓性肌萎缩伴呼吸窘迫1型(spinal muscular atrophy with respiratory distress type 1,SMARD1), 是一种罕见的严重的常染色体隐性遗传性神经肌肉疾病。
大多数SMARD1患儿出生时四肢肌力、 肌张力基本正常,于出生后6周至6个月突然出现膈肌麻痹并呼吸窘迫、呼吸衰竭,四肢肌无力,远端重于近端、下肢重于上肢。
SMARD1的临床表现与脊肌萎缩症有相似点,但发病基因、机制及临床表现有明显区别,临床上对SMARD1认识不足,极易漏诊、误诊。
轴索型腓骨肌萎缩症2S型(Charcot-Marie-Tooth disease, axonal, type 2S; CMT2S; OMIM:616155)是一种以常染色体隐性方式遗传的疾病。患者在10岁左右开始发病,临床症状为进行性远端肌肉无力,远端肌萎缩,脊柱侧弯,马蹄足内翻,远端感觉障碍,步态不稳,腱反射减弱,部分患者舌形异常,轻度近端肌无力。CMT根据神经电生理和病理特点可为不同亚型,最常见的是CMT1 (脱髓鞘型)和CMT2型(轴索型),占所有CMT患者70%-90%;CMTX型占10%-20%,X连锁显性遗传,男性症状严重。

变异解读与讨论
复旦大学遗传咨询在读研究生万远对基因检测报告中提示的变异进行了详细解读。首先通过ClinGen数据库查询到IGHMBP2基因gene curation的结果,IGHMBP2基因导致常染色体隐性远端脊髓型肌萎缩(DSMA1)。
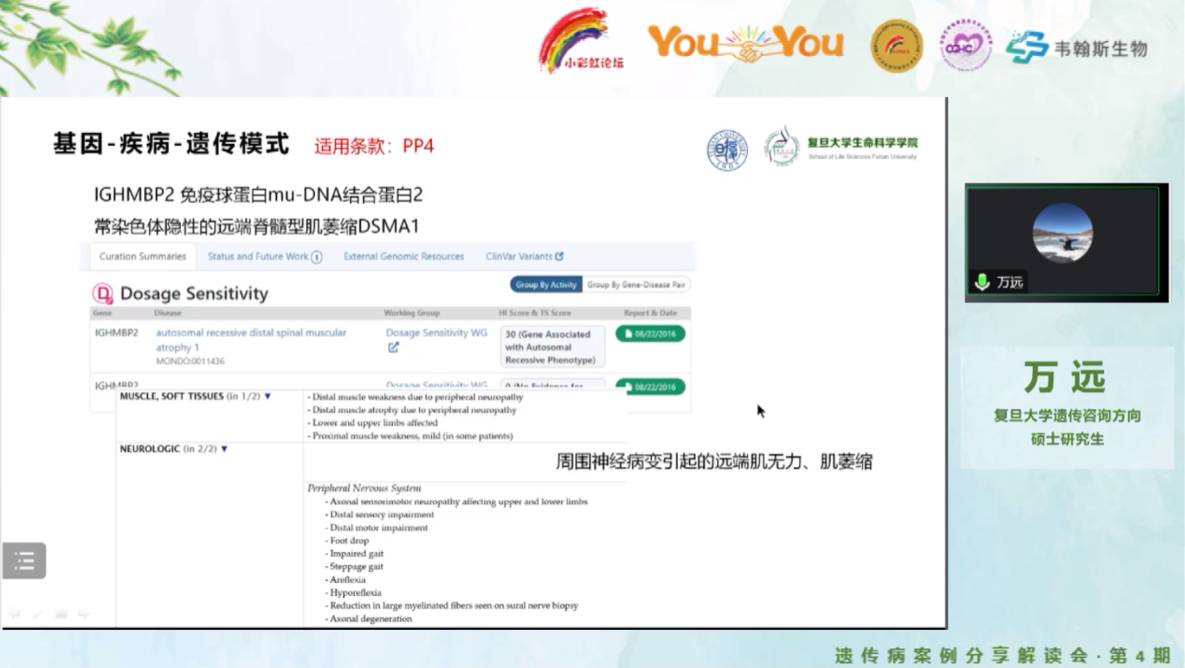
本案例中与临床表型相关的IGHMBP2基因变异c.2362C>T:p.R788*,按照ACMG评级框架,在人群中频率(gnomAD数据库)提示发生罕见符合PM2_Supporting。该无义变异预测是导致IGHMBP2功能丧失,可使用PVS1。IGHMBP2导致远端脊髓型肌萎缩(DSMA1)在OMIM表型中特征表型为“周围神经病变引起的远端肌无力、肌萎缩”,与本例患儿表型较为相符,故使用PP4。另外,该位点在ClinVar上9家单位报道这个变异是致病/可能致病,这个变异是热点变异,查询到文献中存在3个处于反式位置(in trans)的无义变异位点,可以升级为PM3_Strong。最终c.2362C>T:p.R788*评级为致病性变异(P)。
本案例中另外一个位于IGHMBP2基因的变异c.1910G>T:p.R637L,其人群频率在gnomAD数据库中未收录,使用PM2_Supporting。通过分析,PP2(Z score 0.18)、PS1和BP1等都无法适用。ClinVar查询到一个变异具有相同第637位置氨基酸改变c.1909C>T (p.Arg637Cys),ClinVar评估为LP/VUS, 如果是LP,则可以使用PM5。REVEAL预测值为0.743,PP3适用。考虑到与第一个致病性变异位于反式位置,适用PM3。最终第二个变异评级为临床意义不明(VUS)。
附录中另外4个位点,CCDC78基因与中心核肌病4有关,查询发现ClinGen的gene curation结果是limited,SPATA5基因导致伴有听力损失、癫痫发作和脑部异常的神经发育障碍,常染色体隐性遗传方式,表型与本例患儿相符之处较少。LAMA2基因与先天性肌营养不良症1A型相关,主要临床表现为生后早期出现的肌张力低下、运动发育落后、关节挛缩并伴血清肌酸激酶(CK)、肌酸激酶同工酶(CK-MB)等酶学指标升高。89%的变异是LOF变异。本例发现的两个变异位点c.2361A>C和c.8906G>A,根据人群频率、软件有害性预测等证据综合分析,两者仍评为VUS,附录的案例与本案例的临床表型不符合。
合因科技医学服务部总监谭灏文博士结合万远对患儿的变异解读进行点评。
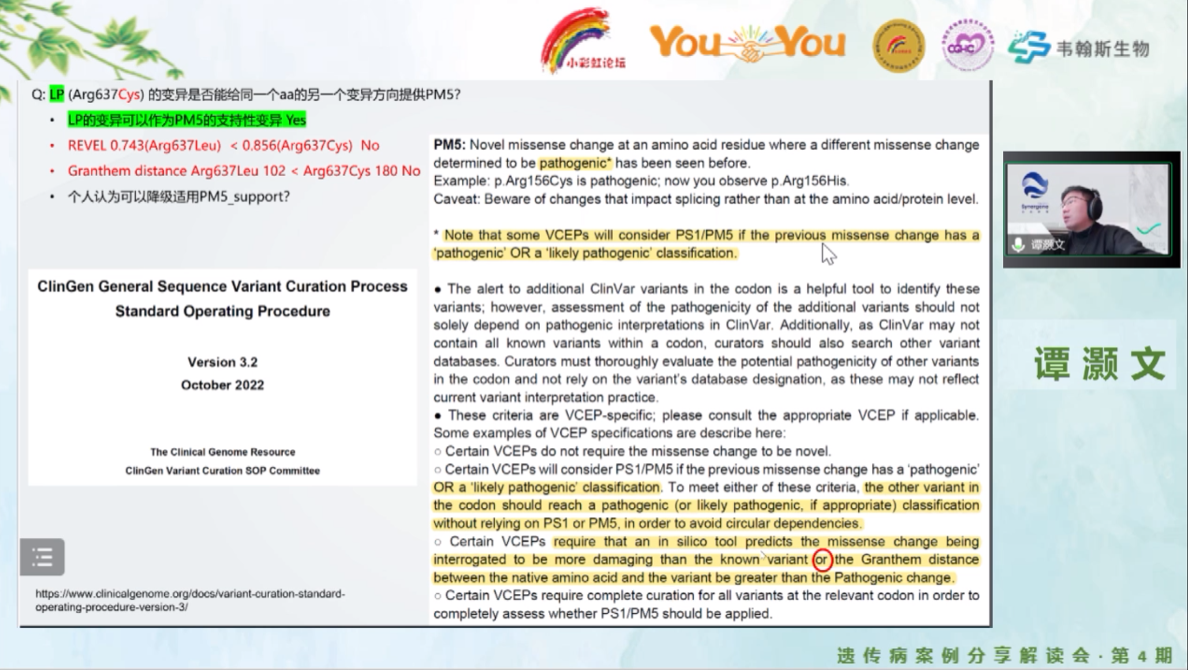
首先IGHMBP2基因变异c.2362C>T是一个东亚热点,报道过多次,评级证据使用的是没有问题的。随后着重讨论了IGHMBP2的另一变异M2,此类错义变异在评级过程中往往难度最大。谭老师认为PP4可以使用,主要考虑患者的症状和辅助检测的结果多吻合,已经通过MLPA检测,排除了腓骨肌萎缩症(CMT)中常见PMP22、MPZ和GJB1基因的拷贝数异常原因,也排除了5q-SMA和DMPK,且进行的是WES检测。对于曾经报道过与第二个变异有相同氨基酸位置的变异p.Arg637Cys,进行了评级:人群频率可使用PM2_Supporting;该点REVEL值为0.856,根据ClinGen最新评分标准,可评为PP3_Moderate;查询文献共找到3个位点与其构成复合杂合,其中2个评为VUS,1个可评为LP,因此可使用PM3评级证据。最终p.Arg637Cys可评到LP等级。由此以来,本案例的第二个变异可以使用PM5证据,ClinGen在2022年10月最新的SOP中,对于使用PM5有新的建议,根据Grandtham distance score,p.Arg637Leu的得分比p.Arg637Cys的得分低,建议降级使用(PM5_Supporting),这样第二个变异升级为可能致病性变异LP。
临床与案例讨论
来自首都医科大学宣武医院的临床神经内科专家王朝东主任结合自身丰富的临床经验,对该案例进行点评。
腓骨肌萎缩症非常常见于成人,主要表现为行走、骨骼和足弓问题。而该病例是一名儿童,相对不太常见。王朝东主任对于PP4的使用提出了临床上的建议。在本案例中,患儿的电生理符合周围神经病变特征。对于在神经科临床做诊断时,建议依据“Midnights”原则对代谢、免疫、感染、营养、中毒等原因进行排查。特别是对于类似没有家族史、父母非近亲的案例,可以增加一些排查性的检查。例如在成人中,遗传性远端型肌病和遗传性周围神经病容易混淆,通过肌酶水平可进行鉴别。另外也可考虑外周神经的活检,如腓肠神经活检。该病非常罕见,获得相关的免疫组化并不容易。但总的来说,临床获得更多的证据,可使得PP4证据更加充分。高永强主任补充,患儿2岁6个月就诊时,腓肠肌萎缩非常不明显。在后续再次见到患儿时,小腿萎缩加重,尚未出现明显的关节变形和足弓异常。过去曾经检查过肌酶是正常范围内。另外,外周神经活检在当地并未广泛开展。在临床实际工作开展中,确实存在一些客观条件的局限性,两位主任的分享与讨论值得大家深入思考。
上海市儿童医院马俐老师在看到本次会议预告后,也主动联系主办方,在本次会议上分享了门诊中遇到的一例IGHMBP2基因突变患儿。女,快2个月,出生时足月小样儿,喂食不多,呼吸等无异样。在当地医院做过常规检查,无明显异常。主任和马俐老师先后均发现女婴吃的不是很好,身上有些松软,活力不好,双脚马蹄足,双手胎儿指垫,但体型不瘦。女婴收治入院后,突然有一天出现呼吸衰竭和酸中毒,只能通过有创呼吸支持,肌无力,远端肌肉重于近端,然而神智清醒。通过家系外显子测序发现该受检者IGHMBP2基因存在一个无义/移码突变和一个单外显子缺失突变,两个位点均未被报道。患儿妈妈回忆曾有过一个出现过类似问题的女儿,送医院后抢救失败。综合临床和分子诊断结果,最终明确该受检者属于SMARD1型。该基因突变案例在国内鲜有报道,感谢马俐老师的分享,该患儿的就诊经历带给了我们一些新的经验。
案例总结
来自复旦大学人类表型组研究院的安宇老师主持各位专家有关该案例的讨论,总结如下:
01
错义变异在临床上通常难以解读,对于错义变异,in trans位点和信息的查询非常重要,能够查询到in trans位点的数量不同可能会使得变异评级的结果大相径庭。UUmatcher等变异位点共享的平台有助于对in trans位点的解读。
02
ClinGen提出了对于PM5新的解读,新的解释较为谨慎。如若使用PM5,首先需满足支持性位点是P/LP,其次还需考虑有害性预测或Grantham distance。在本案例中,安宇老师赞同PM5_Suppoting的使用。
03
本案例临床可能还需要做更多评估,而且随着孩子年龄的增长,也要继续进行随访。不同的基因型和表型的关系还有待进一步关注,不同变异的基因型可能导致表型截然不同。后期可通过持续关注相关报道,不同变异是否出现不同基因型?是否出现无呼吸异常的案例?不同年龄表型是否不一样?还需要分析变异类型与表型的关系。
04
IGHMBP2基因导致常染色体隐性远端脊髓肌肉萎缩 1 型 (DSMA1; 604320 ),也称为脊髓肌肉萎缩伴呼吸窘迫 (SMARD1) 和远端遗传性运动神经元病 VI 型 (HMN6),本案例在儿童期表型未涉及呼吸异常情况,鉴别起来较为困难,通过基因诊断给出了遗传学的检测结果,两个变异都可以评到LP等级。需要继续临床随访,完善相关的鉴别检测,存在的疑问也有待于进一步研究。。
本期小彩虹YOU-YOU“一例儿童遗传性神经肌肉疾病探讨”在大家热烈的讨论与分享中顺利谢幕,专家老师们从临床诊断和遗传变异评级方面做出全面解读和专业点评,不仅使大家对IGHMBP2基因相关疾病有了全新的认识,也对遗传性神经肌肉病有了更深刻的理解和思考。小彩虹YOU-YOU分享会也是一个更加广阔的平台,为各位临床老师和遗传咨询行业人员创造更多获取、分享和交流的机会,欢迎大家来投稿。敬请期待下一期小彩虹YOU-YOU案例解析分享会~
注:本次案例的基因检测由上海韦翰斯生物完成。
本期内容精彩回放请扫描二维码

主办单位:
上海市遗传学会-临床遗传与遗传咨询专业委员会
小彩虹医学遗传学者会
湖北省遗传学会基因健康专业委员会
中国遗传学会遗传诊断分会
全国生育健康遗传关怀合作联盟
上海市遗传病基因检测和出生缺陷三级防控专业技术服务平台
赞助单位:
上海韦翰斯生物医药科技有限公司

上海遗传学会临床遗传与遗传咨询专业委员会旨在以罕见病和遗传病患者为主要关注对象,支持出生缺陷防控咨询师及遗传检测,遗传咨询事业的健康发展,建设基于国际共识和中国国情及患者具体信息和临床需求的权威数据库,知识库和遗传咨询原则,促进临床遗传学,出生缺陷防控及遗传咨询事业发展,为临床医务工作者提供不断更新和长久持续的培训,关注临床遗传和遗传咨询相关研究方向,为遗传咨询相关问题提供科学建议和精准指导。

“小彩虹论坛”-架起医学-基因检测和咨询-遗传机制研究的桥梁
微信扫一扫
关注该公众号
