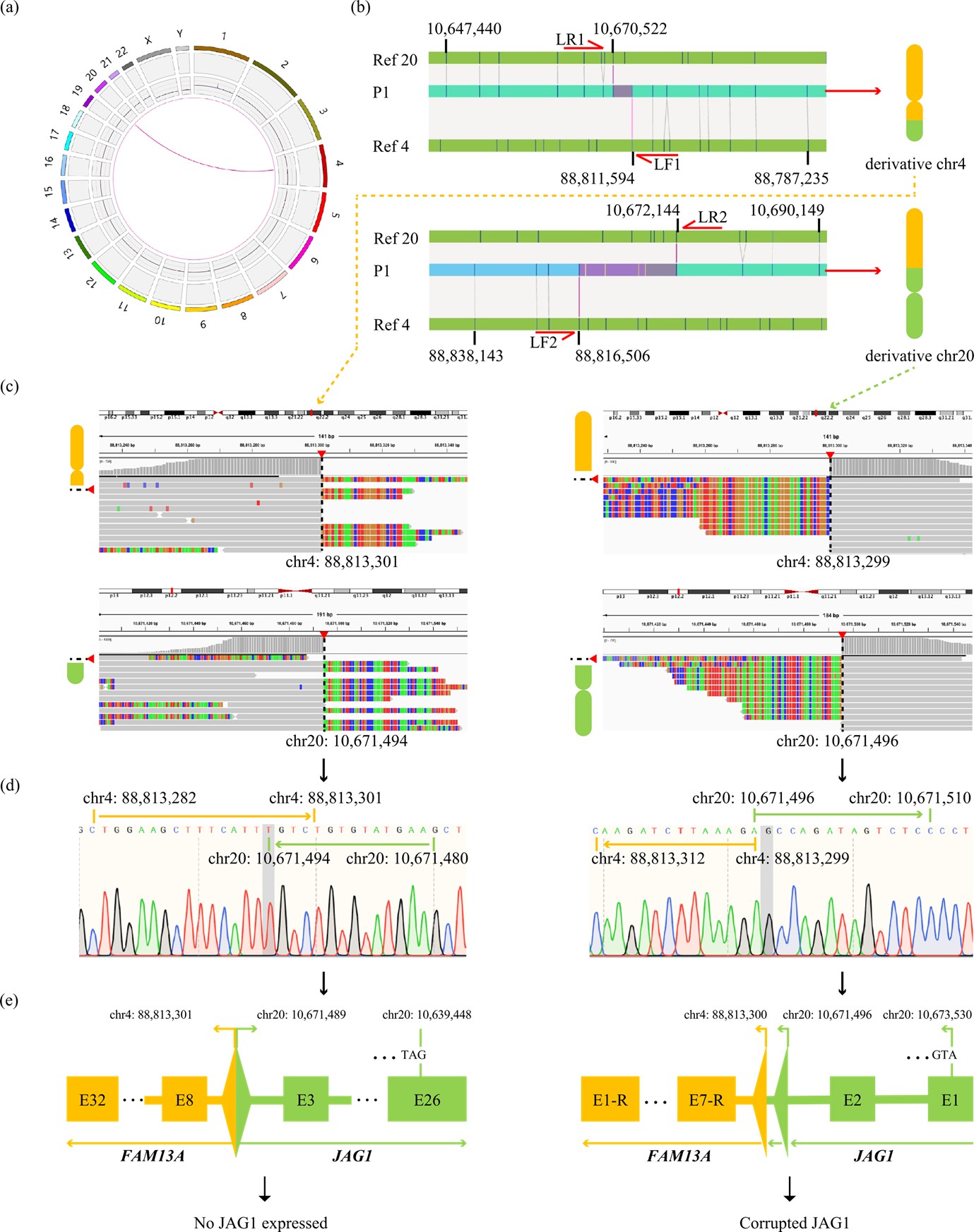
2024年3月,北京大学第三医院血液内科景红梅团队在北大核心期刊《中华血液学杂志》上刊发了一项研究成果。该研究首次利用光学基因组图谱(optical genome mapping, OGM)技术对5例新发的多发性骨髓瘤患者的细胞遗传学异常进行检测。结果显示OGM较传统的细胞遗传学技术能够一次获得更加全面的遗传学信息,节约临床样本,降低检测成本,在多发性骨髓瘤中具有一定的临床应用价值。

本研究所涉及的高分辨率光学基因组图谱技术和染色体微阵列芯片检测均由上海韦翰斯生物医药科技有限公司完成。


一、研究背景
多发性骨髓瘤(MM)是一种以单克隆浆细胞在骨髓中异常增殖为特征的血液肿瘤,其发生率约占所有血液肿瘤的10%。
MM具有复杂的细胞遗传学异常,包括染色体的数目异常和结构变异(SV),它们不仅参与MM的发生、发展,而且在MM
的预后分层体系中占有重要地位。在骨髓FISH检测中,大约40%的多发性骨髓瘤患者的肿瘤浆细胞中存在三体(超二倍体多
发性骨髓瘤),其余大部分会涉及染色体14q32上的免疫球蛋白重链(IgH)位点易位(IgH易位型多发性骨髓瘤)。少部分
患者可同时存在三体和IgH易位。多发性骨髓瘤病程中还会出现其他被称为继发性细胞遗传学异常的细胞遗传学变化,包
括gain(1q)、del(1p)、del(17p)、del(13)和MYC继发性易位。此外,多发性骨髓瘤也存在体细胞变异,常见的突变基因包
括免疫球蛋白重链和轻链基因、NRAS、KRAS和BRAF等。
二、实验方法
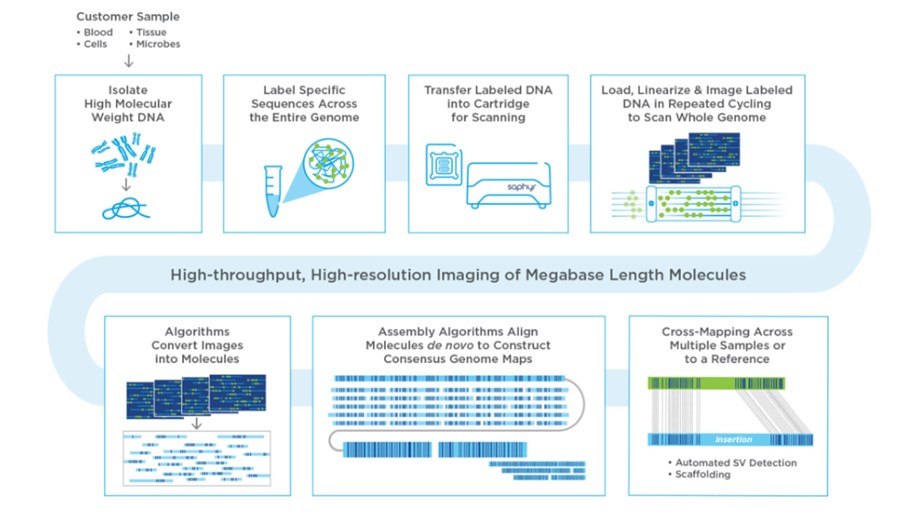
1. 光学基因组图谱(optical genome mapping, OGM):提取骨髓样本中超长基因组DNA, 经特殊位点荧光标记-单分子电
泳-成像-信息转换(图像到分子)-基因组从头组装-SV calling流程获得结构变异数据。此检测及数据分析均由上海韦翰斯生
物医药科技有限公司完成。
2. 染色体微阵列芯片检测:提取骨髓样本基因组DNA,应用 Affymetrix CytoScan 750K 芯片对样本进行全基因组CNV分析
。此检测及数据分析均由上海韦翰斯生物医药科技有限公司完成。
3. 染色体FISH检测:应用磁珠分选技术对骨髓标本中CD138 +浆细胞进行富集,将CD138+浆细胞与相应探针杂交,经洗片
、复染后,在荧光显微镜下观察荧光信号。
4. 染色体核型分析:对骨髓细胞进行24 h培养,常规制片、G显带进行核型分析。
三、实验结果
5例患者OGM检测平均DNA分子长度为227 kb,有效覆盖深度为370X。
与染色体FISH检测能力相比,5例患者FISH检出的染色体异常均被OGM检出,OGM也检出了染色体易位及基因融合上的异常。
与染色体微阵列芯片检测相比,OGM 检出的拷贝数异常的能力显著高于染色体微阵列芯片检测,同时OGM 可以检测平衡易位
,这也是染色体微阵列芯片无法检测的范畴。
与染色体核型分析相比,OGM检出5位患者均存在染色体结构上的变异,但染色体核型分析检测提示有三位受检者核型正常。这
表明MM核型分析容易受骨髓瘤细胞增殖分裂的影响,发生漏检。例4和例5核型结果中虽分别有23%和44%的结构异常没有被
OGM检出,但这些差异结果正确与否还有待证实。


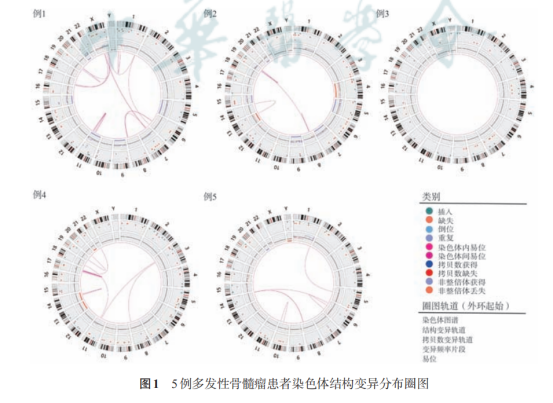
综上所述,OGM较传统的细胞遗传学技术相比,能够一次获得更加全面的遗传学信息,节约临床样本,降低检测成本;而且无需中期分裂相,不受MM细胞体外培养增殖率低的影响,在MM中具有一定的临床应用价值,这些遗传学信息有可能为MM患者进一步危险度分层,评估疾病复发或进展风险提供更多的实验室依据。

OGM技术简介
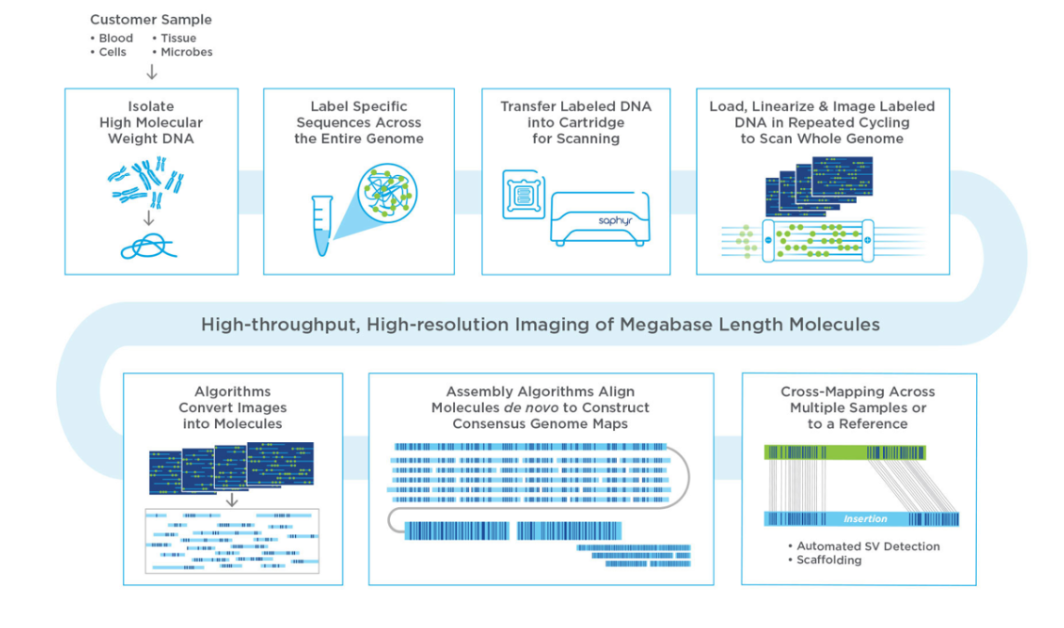
光学基因组图谱技术(optical genome mapping, OGM)是一种新型高分辨率细胞遗传学分析技术,该技术可以利用单个DNA分子基因组限制性内切酶图谱快速生成高分辨率、有序的全基因组限制性内切酶图谱,在检测基因组结构变异的方面具有重大应用价值;这一技术进而可以与现有的高通量测序技术(next generation sequencing, NGS)相结合,优化涉及遗传病、肿瘤、血液病等多个领域的检测流程。

优质服务,助力科研果
韦翰斯面向各类教育研究和医疗机构提供遗传病方向的科研合作,包括基因组学、转录组学、表观遗传学和蛋白质组学四大方向。
在了解到客户的实际需求后,我们经验丰富的管理人员会对项目的需求进行采集和评估,着手制定方案或给出方案建议,还会根
据客户需要进行技术咨询;完成协商、签署合同之后,韦翰斯强大的实验团队会按照合同约定保质保量地完成项目,并且发送包
括质检报告、中期报告和结题报告在内的相关结果,最终再根据客户反馈来妥善安排售后服务。
END
END
一. 研究背景
遗传性球形红细胞增多症(Hereditary spherocytosis, HS)是最常见的遗传性溶血性贫血。临床特征为贫血引起的苍白、高胆红素血症引起的黄疸和脾肿大,黄疸可能是新生儿最重要的体征(通常不存在脾脏肿大)。在中国大陆新生儿中HS的患病率为1:100000,且由于医疗资源不平衡,实际患病率可能更高。HS的临床表现在疾病的遗传和分子基础方面具有高度异质性,虽然实验室诊断中的EMA试验能够有效显示疾病特异性,但没有一项测试可以识别所有遗传性球形红细胞增多症病例。基于二代测序技术的分子检测广泛应用于病原变异的鉴别诊断和确认,可以进一步提高HS诊断的准确性和特异性。
二. 研究对象
王女士(化名)29岁,结婚半年未怀孕,性生活正常且无避孕措施,月经规律,量大无痛,临床检查显示患者血涂片中球形细胞增多,超出正常范围,脾脏肿大,红细胞渗透性脆性增高,胆红素高血症,贫血。根据临床证据,诊断为遗传性球形细胞增多症。为了防止遗传性球形细胞增多症传递给下一代,生殖医学中心的生殖医生、胚胎学家和医学遗传学家建议通过基因检测明确致病机制后,进行胚胎植入前单基因病检测明确胚胎是否遗传致病基因,以实现优生优育。
三. 研究方法
1. 靶向测序
2. 变异位点确认
3. 单细胞全基因组扩增
4. 胚胎植入前全基因组单体型连锁分析
5. 单基因病无创产前诊断
四. 研究结果
对家系成员进行血液疾病相关的panel检测,显示受试者SPTB具有剪接变异(NM_001024858; chr14:65,271,654-65,271,654; exon2; c.300+2dup),经验证父母未携带该变异,表明为新发变异(图1)。

图1
通过Sanger测序结果显示c.279处有低信号峰,及Illumina二代测序结果显示受试者在c.279–299(图2D,红框)处缺失21个碱基(缺少GGTGCTCTGGAGAGATGCT),而在对照样品中未发现缺失,表明c.300+2dup变异影响RNA剪接。
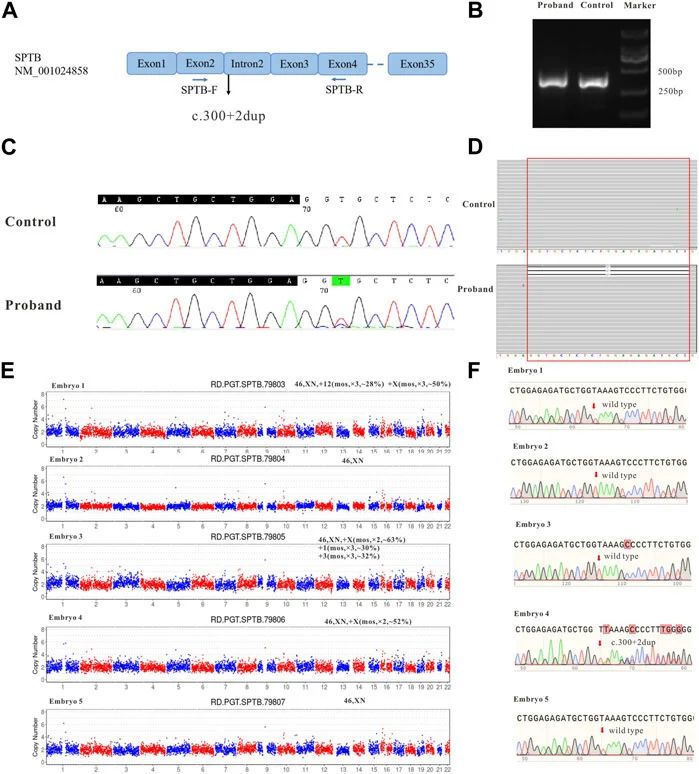
图2
受试者进行体外受精并获得5个囊胚,根据PGT-M和CNV-seq分析的结果,筛选正常核型与不含c.300+2dup变异单倍型的胚胎,最终选择胚胎2进行移植(图2E,F)。移植四周后,B超显示出胎儿心跳,表明受试者成功怀孕。在妊娠13周时,根据图3C所示的程序收集母体外周血进行NIPT-M检测,结果显示胎儿不携带致病变异。受试者在妊娠20周时接受羊膜腔穿刺术,进行产前基因诊断。产前诊断结果(图3D)与植入前基因检测一致,并最终生下没有携带该变异,且出生后所有血液指标均正常的健康男孩。
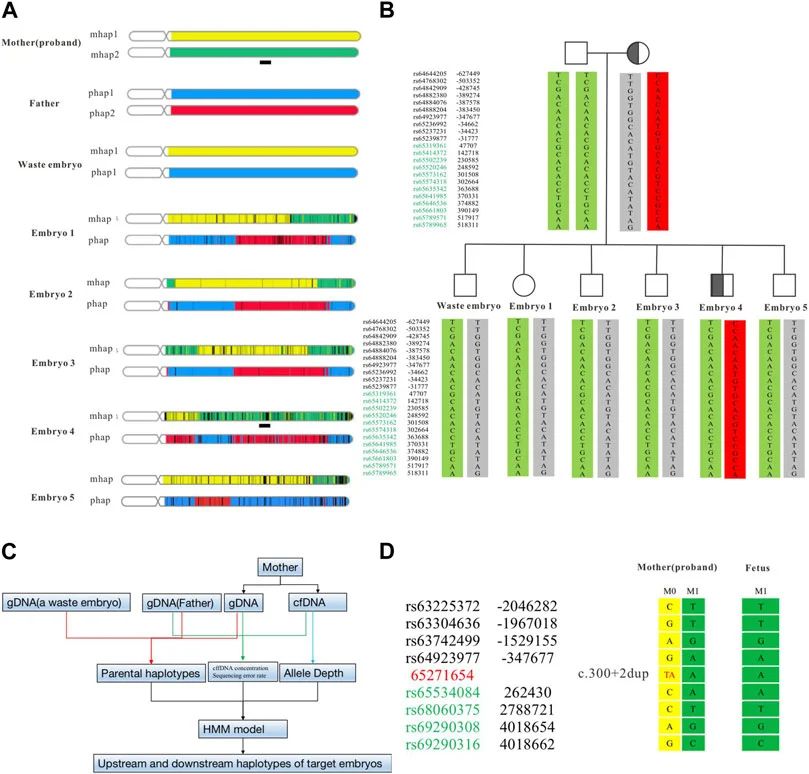
图3
原文链接:https://www.frontiersin.org/articles/10.3389/fgene.2023.1221853/full
注:本文中PGT-M及NIPT-M由上海韦翰斯生物医药科技有限公司提供服务。
PGT-M产品简介:
单基因遗传病PGT-M是经过遗传学检测致病基因,选择不携带已诊断的致病基因变异的胚胎移植,避免子代发病。夫妻一方为单基因病患者或夫妻双方是同一单基因病的携带者、曾孕育或具有生育单基因病患儿高风险的夫妻、及不明原因反复流产或反复种植失败均可以进行PGT-M,阻断单基因病致病基因的垂直遗传以降低出生缺陷发生率。
韦翰斯生物技术优势分析
检测范围广:可检测超过6000种单基因遗传病(SNP、Indel、微缺失等);
准确度高:PGT-A+SNP单倍型分析+一代测序的多重防护;等位基因脱扣率低,数十万个位点可以有效降低等位基因脱扣导致的检测失败和误诊;
检测快速:检测周期短,仅需15个工作日。
蕴可安™不局限于检测几十种显性遗传病,涵盖了2000+基因相关的单基因遗传病,可针对任何有先证者的家系(先证者父母双方或一方为相同致病基因变异的携带者),在极短周期内准确高效检测。在孕早期(怀孕8周)即可检测,准确性>99%,远高于常规产前筛查。目前,韦翰斯生物已取得与国内多家知名三甲医院及医疗机构合作,针对单基因病的无创产前检测,完成数百个家系测试样本的检测分析,其结果与羊水验证结果一致率达100%。
韦翰斯生物技术优势分析
准确可信:准确性>99%,远高于常规产前筛查;测试样本结果与羊水验证结果一致率为100%;
检测范围广:可检测2000+基因,包含1300+显性单基因病和1300+隐性单基因病;
检测周期短:15个工作日;
无创检测:孕妇外周血,非侵入性检测,安全,无感染、致畸或流产风险;
早期检测:孕早期(≥8周)检测,便于后期临床选择;
提供保险:韦翰斯生物为每一位孕妇提供保险,最高可赔付50万。
2023年11月22日,甘肃爱尔眼视光医院|宁夏回族自治区任命医院盛迅伦教授及河南省眼科研究院雷博教授在BMC pediatrics上共同发表了一篇题为《 Exome and genome sequencing to unravel the precise breakpoints of partial trisomy 6q and partial Monosomy 2q》的文章。

该。研究利用全外显子组测序、拷贝数变异测序、三代测序、光学基因组图谱等鉴定了表现骨骼畸形异常、严重智力障碍和严重先天性路脑神经支配失调障碍的患者中存在不平衡易位,并经Long PCR、靶向二代测序和Sanger测序,明确其新发杂合拷贝数缺失和拷贝数重复的精确断点。遗传性疾病的准确分子诊断可以更好地了解复杂综合征的发病机制,这对于有疾病史的家庭和连续异常胎儿表现出相似表型的夫妇来说是必不可少的。
一、研究背景
一位5岁的女患儿因双侧上睑下垂入院,经过全面的眼科检查,发现女孩眼睑裂向下倾斜和双侧视神经发育不全,面部畸形包括突出的前额、中脸发育不全、眼距过大、鼻梁扁平、鼻子短、嘴唇薄的小嘴、小颌畸形和眼睑包茎;产后生长障碍,后发展为严重的智力障碍;并表现严重的神经和骨骼畸形异常,胸部发育异常及先天性颅脑神经支配失调障碍。
该女陔母亲有两次流产史,女孩的父母和她的姐姐(10岁)都很健康。女孩的叔叔表现出类似的表型:面部畸形、胸部和肋骨短畸形,并在35岁时死于肺炎。
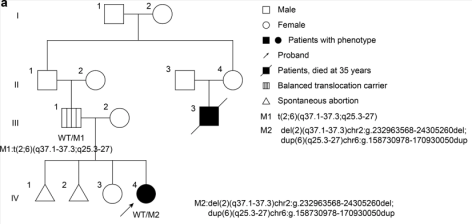
(家系图)
二、研究方法
Ø 全外显子组测序 (Whole-exome sequencing,WES)
Ø 拷贝数变异测序 (Copy number variation sequencing,CNV-seq)
Ø 核型分析(Karyotype analysis)
Ø 三代测序(Oxford nanopore sequencing,ONT)
Ø 光学基因组图谱 (Optical genome mapping,OGM)
Ø 断点验证:靶向二代测序 (Targeted next-generation sequencing) +Sanger 测序(Sanger sequencing)
三. 研究结果
1.WES提示受检者存在拷贝数变异(缺失/重复)
受检者表型复杂,临床建议基因检测明确病因,因此对先证者、姐姐和父母进行了家系WES,提示了先证者存在两个拷贝数变异,一个是2号染色体的杂合缺失,另一个是6号染色体的杂合重复。CNV-seq结果验证变异真实存在,即6q25.3-q27区域有12.01 Mbp重复,2q37.1-q37.3区域有9.23 Mbp缺失。

2.先证者的父亲存在平衡易位
核型分析显示先证者的核型为46,XX,del(2)t(2;6)(q37.1;q25.3),其母亲为正常的 46,XX 女性核型,其父亲的核型为46,XY,t(2;6)(q37;q25.3),父亲在 2q37 和 6q 之间具有平衡的相互易位。
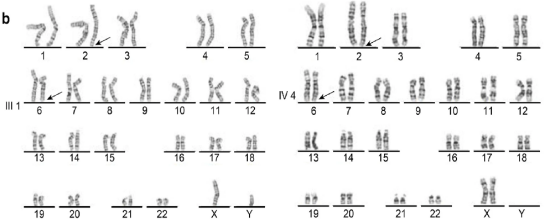
3.断点验证
ONT和OGM证实了先证者中存在2q37.1-37.3处的异常缺失以及Chr2和Chr6的染色体重排,鉴于先前的结果,进一步利用Long PCR靶向二代测序和Sanger测序明确了先证者中的杂合拷贝数缺失 del(2)(q37.1q37.3)chr2:g.232963568_24305260del 和拷贝数重复 dup(6)(q25.3q27)chr6:g.158730978_170930050dup的断点位置。
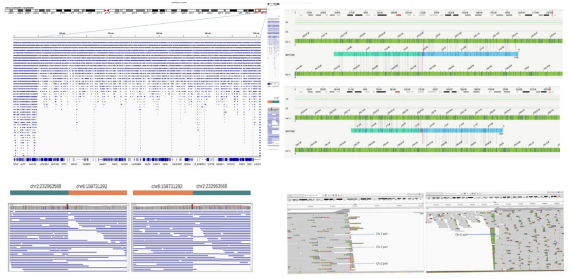

注:本文中实验技术均由上海韦翰斯生物医药科技有限公司提供服务。
OGM技术简介
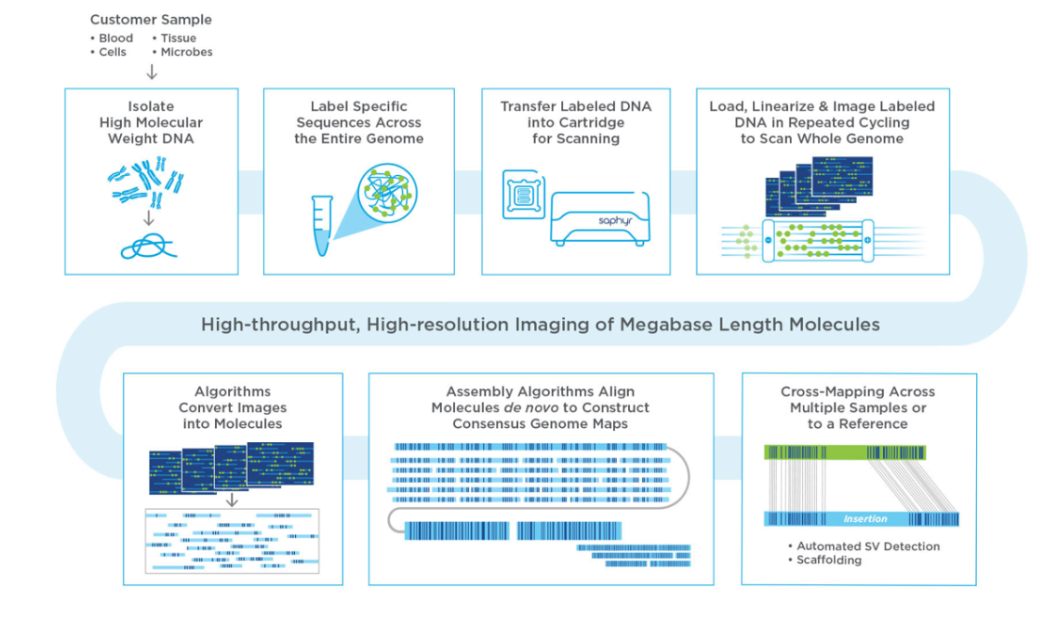
光学基因组图谱技术(optical genome mapping, OGM)是一种新型高分辨率细胞遗传学分析技术,该技术可以利用单个DNA分子基因组限制性内切酶图谱快速生成高分辨率、有序的全基因组限制性内切酶图谱,在检测基因组结构变异的方面具有重大应用价值;这一技术进而可以与现有的高通量测序技术(next generation sequencing, NGS)相结合,优化涉及遗传病、肿瘤、血液病等多个领域的检测流程。
NGS+OGM
韦翰斯全外显子组测序(WES)覆盖20,000+人类功能核基因(含毗邻区域)和线粒体基因组,可高效实现大部分遗传病患儿的精准诊断。随着结构变异的研究深入,中等大小的基因组复杂结构变异在临床遗传病中占到难以预估的比例。
通过光学基因组图谱技术(OGM)结合WES,可实现对人类基因组多种变异类型的全面覆盖,不仅可以提高临床的检出水平,同时有望发现新的结构变异与疾病间的关系,从而扩展科研思路。

_NEWS_
高分辨率光学基因组图谱在检测疑难罕见遗传性肝病中的临床应用
Human mutation
2023年6月8日,遗传学经典期刊Human mutation (Q2区,IF=4.7, Web of Science 收录)在线刊发了复旦大学附属儿科医院肝病科王建设教授团队一项研究成果。本研究对一例临床确诊阿拉杰里综合症,多重连接探针扩增技术(multiplex ligation-dependent probe amplification, MLPA)、基因Panel和全基因组测序(whole genome sequencing,WGS) 结果均为阴性的先证者进行光学基因组图谱(optical genome mapping, OGM)检测,发现4号和20号染色体上一个平衡易位变异,经Long-range PCR、靶向二代测序和Sanger测序,确定了4号和20号染色体断裂位点。该变异破坏了JAG1基因结构,使其无法表达出有正常功能的JAG1蛋白 。
本研究结果证明了OGM对染色体相互易位变异的高分辨率检测能力,推荐作为疑难遗传性肝病检测的一线检测手段。”上海韦翰斯生物医药科技有限公司提供OGM检测和断点验证服务。韦翰斯生物高鹏飞先生作为共同第一作者。

一、研究背景
二、实验方法
三、实验结果
原文链接
https://www.hindawi.com/journals/humu/2023/5396281/

扫码阅读原文

OGM 技术简介
优质服务,助力科研果
2022年7月28日,免疫学经典期刊Journal of Clinical Immunology(Q1区,IF=8.5)在线刊发了复旦大学附属儿科医院临床免疫/过敏科一项研究成果。本研究对一例临床确诊肉芽肿,全外显子组测序(whole exome sequencing,WES)只找到一个NCF2杂合致病变异(来自母亲)的先证者进行光学基因组图谱(optical genome mapping, OGM)检测,发现一个~1500bp NCF2杂合缺失致病结构变异,经Long-range PCR、二代测序和Sanger测序,明确了该结构变异(来自父亲)。本研究结果证明了OGM对 >500bp 插入/缺失型结构变异和杂合型变异的高分辨率检测能力,推荐作为疑难肉芽肿案例基因结构变异检测的一线检测手段。”上海韦翰斯生物医药科技有限公司提供OGM检测和断点验证服务。韦翰斯生物CTO杨敬敏博士作为本文共同第一作者。

研究背景
慢性肉芽肿病(chronic granulomatous disease, CGD)是NADPH氧化酶复合物功能缺陷导致的原发性免疫缺陷病。患者因吞噬细胞无法正常产生超氧离子、过氧化氢来杀伤细菌、真菌、结核等病原,导致出现早发、反复的感染。致病基因为CYBB、CYBA、NCF1、NCF2、CYBC1、NCF4。临床发现,95%以上的致病变异为单核苷酸变异(single nucleotide variants,SNVs)。研究发现,拷贝数变异(copy number variants, CNVs)和结构变异(structural variants, SVs)也是CGD不可忽视的致病原因。本研究借助光学基因组图谱技术,明确了一例CGD患者的隐匿致病性CNVs/SVs。
实验方法
1. 光学基因组图谱(optical genome mapping, OGM):从先证者外周血中抽提 >150 kb长度的基因组DNA, 经特殊位点荧光标记-单分子电泳-成像-信息转换(图像到分子)-基因组从头组装-SV calling流程获得结构变异数据。
2. 结构变异分析解读:根据变异置信度、变异的人群频率、与表型的相关性等找到致病变异。
3. Long-range PCR和二代测序, 用以明确变异断点在染色体上的位置。
4. Sanger测序,进一步确认断点和变异来源。
实验方法
OGM在一例肉芽肿患者基因组中发现NCF2基因存在一个杂合片段缺失,缺失长度为1,506bp ~ 1,526bp。缺失片段的上游位于4个胸腺嘧啶(T)位置,下游位于胸腺嘧啶(T)富集区域(参考序列为18个T,P1为58个T,多的T由DNA双链断裂修复机制产生)。该变异来源于父亲。
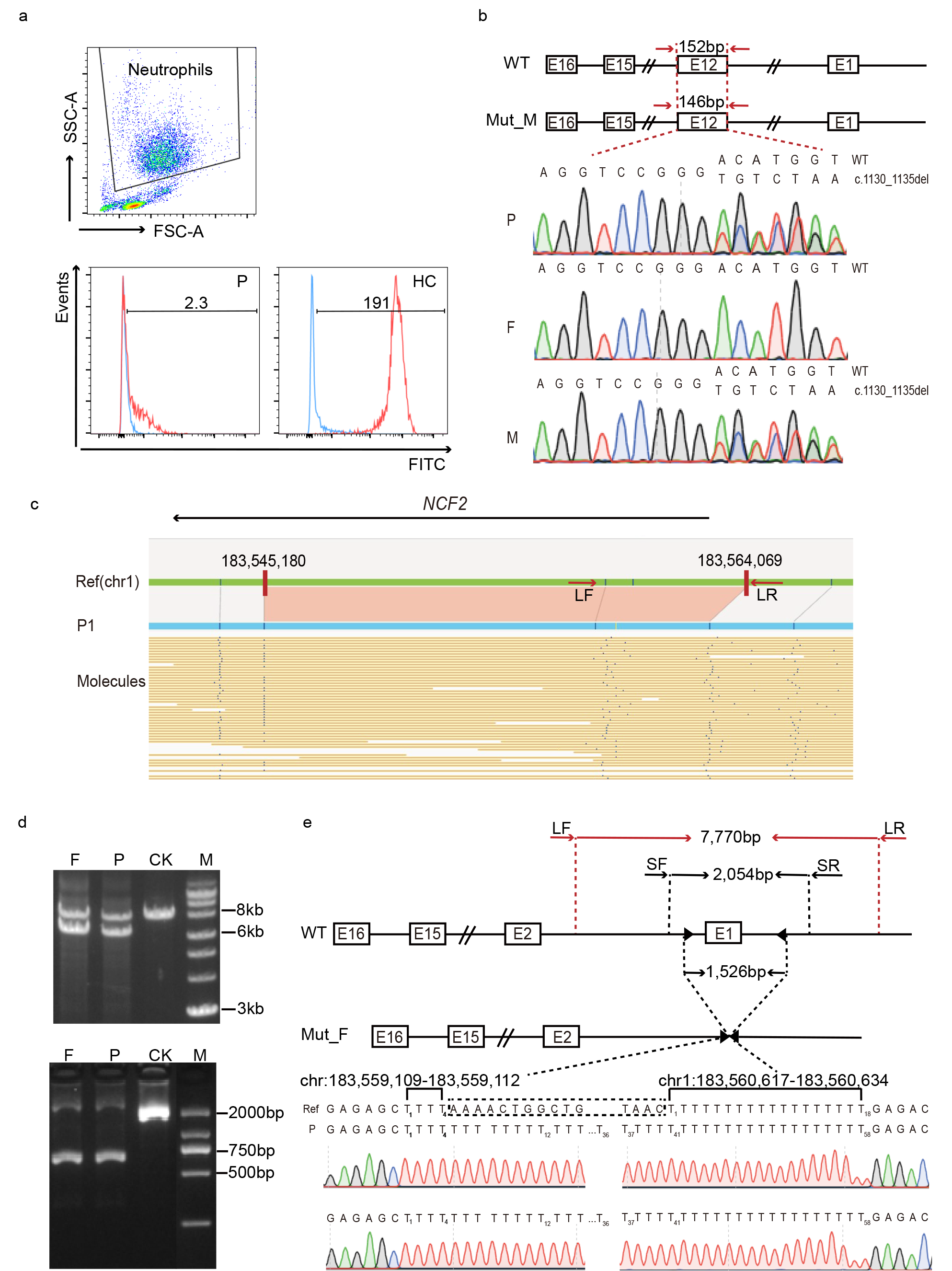
原文链接 https://pubmed.ncbi.nlm.nih.gov/35900637/
OGM 技术简介
光学基因组图谱技术(optical genome mapping, OGM)是一种新型高分辨率细胞遗传学分析技术,该技术可以利用单个DNA分子基因组限制性内切酶图谱快速生成高分辨率、有序的全基因组限制性内切酶图谱,在检测基因组结构变异的方面具有重大应用价值;这一技术进而可以与现有的高通量测序技术(next generation sequencing, NGS)相结合,优化涉及遗传病、肿瘤、血液病等多个领域的检测流程。
优质服务,助力科研
韦翰斯面向各类教育研究和医疗机构提供遗传病方向的科研合作,包括基因组学、转录组学、表观遗传学和蛋白质组学四大方向。
在了解到客户的实际需求后,我们经验丰富的管理人员会对项目的需求进行采集和评估,着手制定方案或给出方案建议,还会根据客户需要进行技术咨询;完成协商、签署合同之后,韦翰斯强大的实验团队会按照合同约定保质保量地完成项目,并且发送包括质检报告、中期报告和结题报告在内的相关结果,最终再根据客户反馈来妥善安排售后服务。


近日,医学遗传学经典期刊Journal of Medical Genetics(Q1区)在线刊表了复旦大学附属妇产科医院集爱中心一项研究成果。该研究通过对多种结构变异检测技术在隐匿性平衡染色体重排(Balanced Chromosomal Rearrangements, BCRs)的检测比较,揭示了高分辨率光学基因组图谱(optical genome mapping, OGM)在隐匿性染色体重排变异检测中具有独特优势,并推荐可以将OGM作为一种新的检测隐匿性BCRs的临床一线手段。
研究背景
染色体重排在多种人类遗传疾病发生发展中有着深远的影响,BCRs变异携带夫妇发生流产或生育缺陷患儿的风险异常升高。临床上,根据染色体易位片段大小和易位区带条带特征可分为肉眼可见的和隐匿性的BCRs,其中前者可通过常规染色体核型分析明确诊断,而后者却超出核型分析的检测能力。当前荧光原位杂交(Fluorescence in situ hybridization,FISH)技术可以诊断BCRs,但前提是需要通过CMA或CNV-seq在携带者夫妇流产组织或受累子代中检测到染色体不平衡重排作为提示,且需要位点特异性荧光探针及中期细胞分裂相培养,所以FISH的临床应用限制较大。因此,临床上需要探索一种更加高效、便捷且准确性高的检测BCRs的新技术方法。
研究路线


研究对象与结果
该研究通过对11对染色体核型正常但有复发流产或受累后代的夫妇进行OGM基因组范围内的染色体结构变异检测,在受检者中检测到不同类型的染色体重排事件,其中包含隐匿性的相互易位、小片段插入及倒位连接的易位等,同时检测结果还提示了重排染色体的较精确的断点区域。基于OGM提示的断点区域,研究者们通过LR PCR+Sanger测序进一步确定了变异的真实性和精确断点。此外,研究还通过CMA芯片检测、CNV-seq和FISH技术证实了变异的准确性。
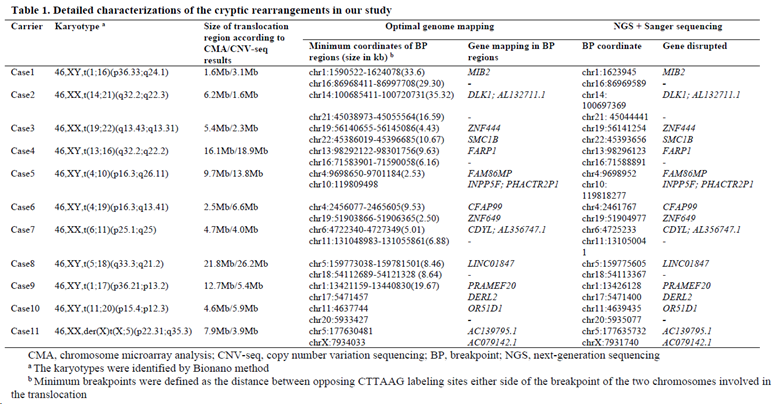
(受检者中隐匿性重排的变异详情)

(OGM变异检测结果及LR PCR和Sanger测序验证结果)
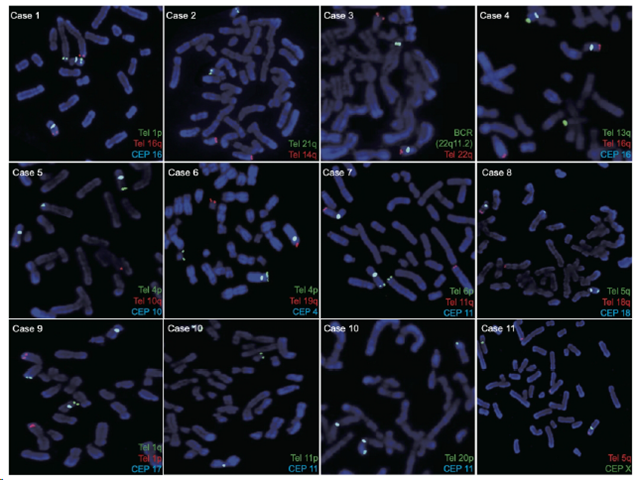
(FISH变异验证结果)
研究结果表明,OGM技术能够准确检测到隐匿性BCRs,且可以精准定位出断裂点所在区带,结果准确可靠。此外,OGM可以揭示额外的复杂重排事件,能进一步细化相关潜在的遗传解释。该研究结果提示OGM在诊断隐匿性BCRs方面具有独特的优势,可作为临床常规检测的一线方法,为BCRs临床诊断提供了新思路、新视野。该研究的实验部分由上海韦翰斯生物提供。
OGM技术简介
光学基因组图谱技术(optical genome mapping, OGM)是一种新型高分辨率细胞遗传学分析技术,该技术可以利用单个DNA分子基因组限制性内切酶图谱快速生成高分辨率、有序的全基因组限制性内切酶图谱,在检测基因组结构变异的方面具有重大应用价值;这一技术进而可以与现有的高通量测序技术(next generation sequencing, NGS)相结合,优化涉及遗传病、肿瘤、血液病等多个领域的检测流程。
优质服务,助力科研
韦翰斯面向各类教育研究和医疗机构提供遗传病方向的科研合作,包括基因组学、转录组学、表观遗传学和蛋白质组学四大方向。
在了解到客户的实际需求后,我们经验丰富的管理人员会对项目的需求进行采集和评估,着手制定方案或给出方案建议,还会根据客户需要进行技术咨询;完成协商、签署合同之后,韦翰斯强大的实验团队会按照合同约定保质保量地完成项目,并且发送包括质检报告、中期报告和结题报告在内的相关结果,最终再根据客户反馈来妥善安排售后服务。
今天跟大家分享一篇于2022年4月28日刊载在Frontiers in Pediatrics期刊(IF:3.418)上的病例报道,由成胜权教授和陶东英医生等人撰写,来自韦翰斯的杨敬敏博士参与了该项研究,先证者是一名20个月大的丙酮酸羧化酶缺乏症(PCD)患儿,系国内首个报道的PCD病例,文章探明了该患儿的新型剪接突变,并证明了其致病性;不但填补了国内PCD相关临床信息的空白,也丰富了PC基因的突变谱系,为该病的诊治贡献了新的思路。
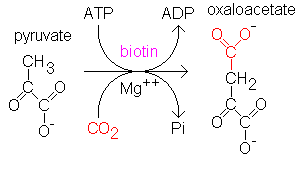
丙酮酸羧化酶(PC)作用示意图:丙酮酸羧化酶(pyruvate carboxylase)是一种线粒体基质酶,可将丙酮酸转化为草酰乙酸(oxaloacetatic acid),进而参与糖异生和能量产生的过程;
PC活性降低将导致:
①丙酮酸的积累,随后在体内转化为乳酸,最终引发乳酸性酸中毒 ;
②草酰乙酸的减少还会使糖异生发生减少,导致机体供能不足;
③其他相关的临床表型还包括酮症酸中毒和高氨血症等。
丙酮酸羧化酶缺乏症简介
丙酮酸羧化酶缺乏症(PCD)是一种罕见的常染色体隐性遗传疾病,由丙酮酸羧化酶(PC)的活性不足所致,其最为常见的临床特征包括神经发育迟缓、丙酮酸水平升高、乳酸性酸中毒、酮体水平升高和高氨血症。目前PCD主要表现为三种临床形式:
婴儿型(A型),主要在北美洲人群中发现。通常于患者出生几个月之内(婴儿期)发病,典型特征包括肌张力减退、发育迟缓和乳酸血症,随后出现感染、腹泻和其他症状,最终导致婴儿期或儿童早期死亡;
新生儿型(B型),主要在欧洲发现,尤其以法国人群为主。通常以严重的乳酸酸中毒、高氨血症为特征,患者通常于出生后3个月内死亡;
迟发型(C型)病情较轻,也称为温和型。通常表现为正常或轻度延迟的神经发育和发作性代谢性酸中毒。
截止文章成稿之时,全球范围内仅报告了33例PCD病例,而且没有一例发生在中国人群,因此本文描述的先证者为国内首例PCD患儿。
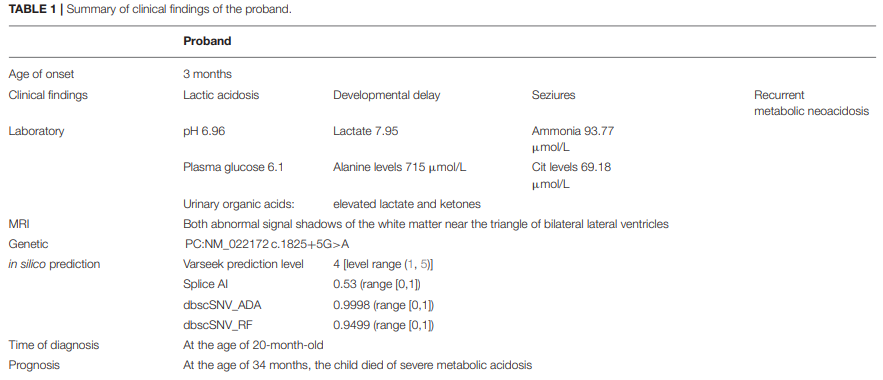
先证者情况介绍
先证者是一名20个月大的男童,是一对身体健康、近亲结婚夫妻的第一个孩子,最开始因为精神发育迟缓和反复乳酸酸中毒入院。患儿足月出生,初始的身材体型均属正常,但在3个月时出现双侧眼睑阵挛发作,每次持续30秒到1分钟;成长至9个月大时,又出现了严重的乳酸性酸中毒,表现出生长迟缓、呼吸急促和嗜睡等症状,急性发作期间还会出现一系列实验室检查指标上的异常:血浆pH值降低、血浆乳酸/血浆氨和葡萄糖以及丙氨酸和瓜氨酸水平显著增高;随后在患儿34个月大时,又出现了一次极为严重的代谢性酸中毒发作,尽管进行了透析治疗,患儿的病情仍然持续恶化,乳酸水平从7.7 mmol/L上升至23.4 mmol/L,最终不治身亡。
明确致病突变
尽管先证者的生命已经无法挽回,研究者还是需要将这个宝贵的案例研究透彻,以明确国内首例PCD患儿的致病原因。通过观察患儿的临床症状已经可以初步判断为婴儿型(A型)丙酮酸羧化酶缺乏症,随后研究者通过家系全外显子组测序(WES)的手段确认了诊断;进一步的发现表明,先证者是PC基因中(c.1825+5G>A)突变的纯合子,该位点的突变在Genome Aggregation Database (gnomAD) 或1000 Genomes 数据库中均未有过报道。
随后为了验证该突变位点的致病性,研究者使用四种不同的预测软件(Varseak、splice AI、dbscSNV_ADA和dbscSNV_RF)对该突变的影响进行预测后,结果一致表明其会导致PC基因mRNA的异常剪接;
紧接着研究者对先证者及其父母,还有健康对照组的cDNA扩增产物进行了NGS测序,证实在先证者的cDNA中保留了部分PC基因13号内含子结构,从而导致蛋白编码异常,而父母双方都是相同异常剪接突变的杂合携带者;
最终使用Sanger测序与健康对照进行对比验证后,得出了可靠的结论:PC :c.1825+5G>A的突变会引起丙酮酸羧化酶基因mRNA的异常剪接,先证者样本中保留了192 bp的13号内含子结构,根据ACMG指南该变异被评定为致病;综合考虑临床表现、基因分析和cDNA测序结果,研究者最终确定该突变为患者PCD的病因。
图A:体内RT-PCR片段的凝胶电泳显示,先证者的条带大于对照组,证明先证者的cDNA可能存在异常剪接;
图B:对cDNA的NGS测序结果显示,先证者的cDNA保留了部分PC基因的13号内含子且为纯合子,其父母双方均为杂合子;
图C、D:PC基因的示意图,显示出了剪接突变发生的位置以及Sanger测序结果,表明该剪切突变导致PC基因的13号内含子发生了192bp的保留。
早确诊,早治疗
对于该病的鉴别诊断,需要注意与其他遗传代谢病——尤其是容易导致高乳酸血症和神经发育异常的类别进行区分,例如I 型糖原积累综合征 (GSD1)、丙酮酸脱氢酶复合物缺乏症 (PDHCD)、遗传性果糖不耐受和果糖-1,6-双磷酸酶缺陷等等。在此过程中,一条重要的思路是发现其他疾病包括的,但是PCD表型谱中没有的一些症状,比如常见于 I 型糖原积累综合征和果糖-1,6-双磷酸酶缺乏症中的低血糖和肝肿大;或者像PDHCD与PCD在临床表型上虽然没有差异,但无法在PDHCD患者中检测出血酮体。
但是PCD本身就是极为罕见的疾病,更何况国内临床上对PCD的认知尚处于经验不足的状态,再考虑到遗传代谢病通常具有复杂的表型异质性和外显率的变化,单单通过对于患者症状的观察即使可以做到大致上的判断视其为疑似病例,但最终出于严谨和负责的角度还是有必要从基因层面进行确诊——正如文献中的研究者所做的那样。
本文报道的病例不仅是国内首例PCD病例,其突变位点还是首次发现的新型致病变异,对于该剪接突变致病性的证明不仅丰富了PC基因的突变谱系,也为此后该类疾病的基因诊断提供了新的依据。
文中提到的患儿于三个月大时表现出临床症状,初期的表现并没有呈现出很强的特异性,直到后续随着病情进展出现严重的发育迟缓和代谢异常,才引起了家属的重视送往医院治疗,但最终还是无力回天。虽然针对早期症状就做出准确的诊断困难重重,但其实并不难判断患儿的症状是由遗传病引起的,如果能够在早期就明确病因、进行治疗,多数疾病都会有更好的治疗和预后效果。
韦翰斯全外显子测序项目
(WES)
韦翰斯的全外显子测序项目(WES)可以一次检测人类基因组中近2万个已知核基因的外显子和毗邻剪接区域(±20bp),以及线粒体基因组全长测序,可同时检测线粒体突变导致的相关疾病,是针对病因不明的遗传病患者最全面、高效的解决方案。无论是发病率极低的罕见病,还是表型缺乏特异性难以诊断的遗传病,甚至像文中患儿这般是由于尚未收录的突变致病的情况,都可以通过WES的检测手段发现可能的致病突变,再经由韦翰斯认真负责的生信分析及人工解读去出具一份详实可靠的报告,把真相和希望带给患者和他们的家属。
优质服务,助力科研
韦翰斯面向各类教育研究和医疗机构提供遗传病方向的科研合作,包括基因组学、转录组学、表观遗传学和分子生物学四大方向。在了解到客户的实际需求后,我们经验丰富的管理人员会对项目的需求进行采集和评估,着手制定方案或给出方案建议,还会根据客户需要进行技术咨询;完成协商、签署合同之后,韦翰斯强大的实验团队会按照合同约定保质保量地完成项目,并且发送包括质检报告、中期报告和结题报告在内的相关结果,最终再根据客户反馈来妥善安排售后服务。
“
慢性进行性眼外肌麻痹(CPEO)是一种慢性进行性上睑下垂及眼球运动障碍,双侧眼外肌均受累,且眼肌麻痹极为缓慢,复视较少见。部分患者可合并四肢肌无力和运动不耐受。
CPEO也是线粒体大片段缺失突变中较为常见的表型,少数患者可检测到mtDNA的点突变(A3243G、G12316A、T4274C)。任何年龄均可发病,诊断年龄以41岁以上居多。
线粒体DNA缺失综合征包括三个临床表现相关的综合征:Kearns-Sayre综合征(OMIM # 530000),进行性眼外肌麻痹(OMIM #157640)和 Pearson综合征(OMIM # 557000)。这三种综合征的症状可以在同一家庭中的病人里,或同一病人不同的疾病期发生转换。
除了CPEO以外,KSS和Pearson综合征也主要为mtDNA大片段缺失突变导致的临床表型,其发病机制相似,临床症状取决于mtDNA缺失在不同组织中的分布,线粒体的分布也存在组织异质性,即不同的组织线粒体分布数量不同。
因此线粒体疾病在进行mtDNA基因突变分析时应选择合适的检测方案,特别注意标本的选取,关注不同组织细胞中异质性比例。
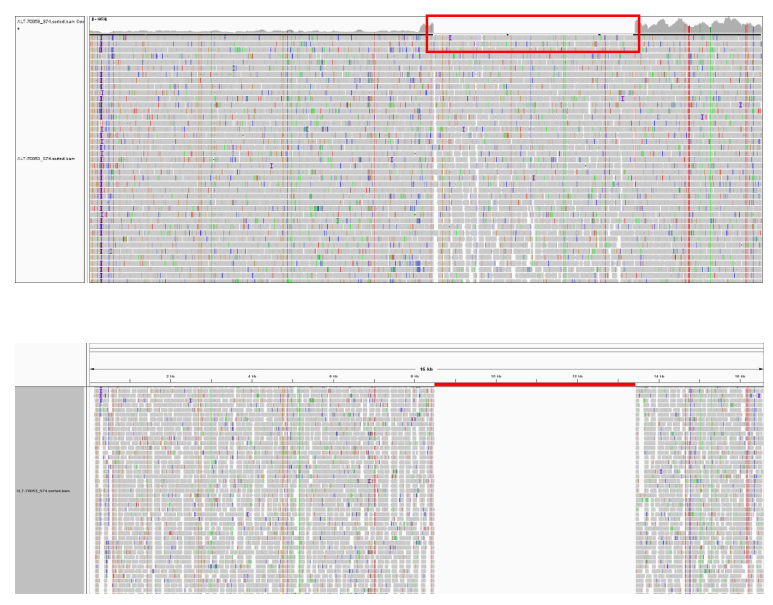
案例1 :
其他机构采了患者的外周血,做眼科的panel,仅发现受检者线粒体基因组的chrM-11084有单个碱基突变,该位点为临床意义未明。
检测数据经和正常人线粒体基因组结果比对,在基因组水平未发现大片段的缺失和重复。
对受检者进行高通量测序检测线粒体 DNA 全部序列,发现受检者 m.****-***** 存在约9,122 bp 缺失,
该变异可能导致线粒体遗传的 Kearns-Sayre 综合征(KSS, OMIM #530000)。

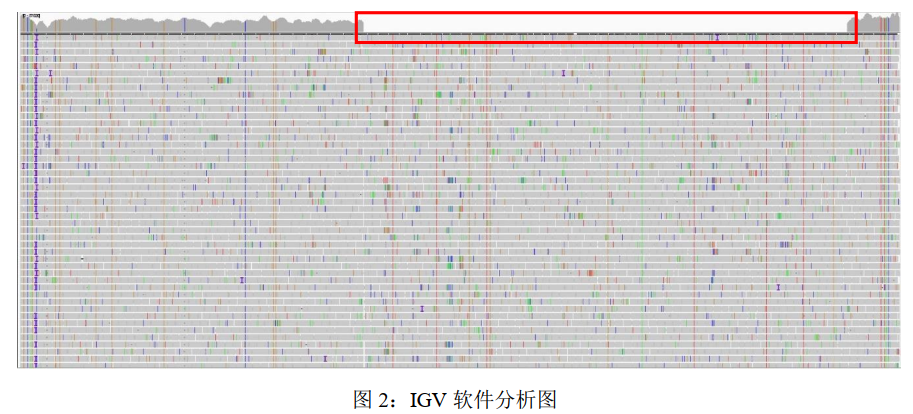
Kearns-Sayre 综合征是一种线粒体缺失综合征,有组织异质性,可能会导致组织和血液的异质性比例不一致的情况,
现在组织的高深度测序显示,异质性变异比例98%以上,与电泳图结果一致。
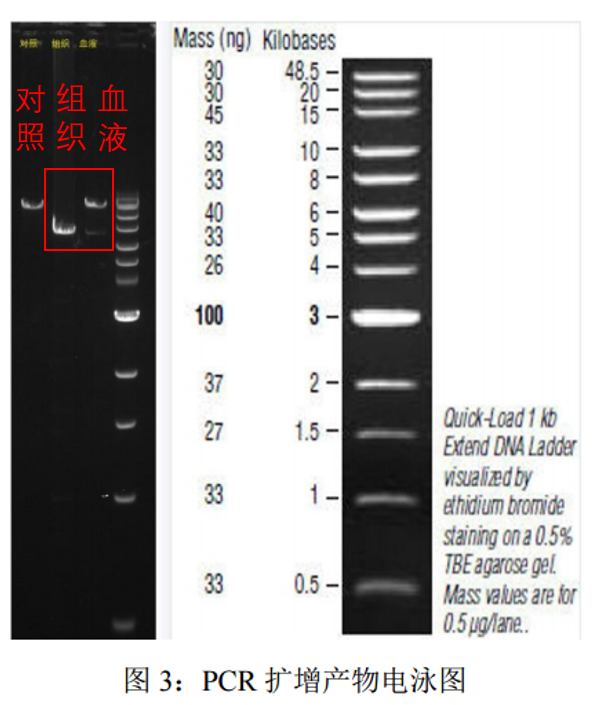
案例2:
对受检者进行高通量测序检测线粒体 DNA 全部序列,发现受检者 m.****-***** 存在约4964bp 缺失,
该变异可能导致线粒体遗传的 Kearns-Sayre 综合征(KSS, OMIM #530000)。

CPEO、KSS综合征等线粒体缺失综合征,应根据患者的具体的临床症状,注意受检者的送检样本类型和基因检测方案的选择,综合考虑线粒体的组织异质性,来更好的提高线粒体疾病的检出率。
疾病概述
慢性进行性眼外肌麻痹(CPEO)是以上睑下垂为首发症状的一种遗传性眼病。临床表现为慢性进行性上睑下垂及眼球运动障碍,因为双侧眼外肌均受累,且眼肌麻痹极为缓慢,所以复视较少见。部分患者可合并四肢肌无力和运动不耐受。
以往的研究表明本病的发生与线粒体(mtDNA)的损伤有关,属于线粒体脑肌病的一种类型。
随着基因检测技术的发展,
mtDNA 大片段缺失、点突变及与线粒体功能密切相关的核基因突变进一步证实CPEO 及 CPEO 叠加综合征是一类线粒体疾病。
疾病介绍
CPEO
临床表现
CPEO是一种线粒体肌病,表现为进行性眼外肌麻痹,即上睑下垂和眼球活动障碍,双眼可同时或先后发病,瞳孔可正常,复视少见。
病情缓慢进展,眼外肌可逐渐纤维化,最终可出现眼球固定,可伴有视网膜病变、视神经萎缩、角膜浑浊等,部分患者存在四肢近端肢体无力。
临床诊断
肌活检病理可见破碎红纤维(ragged red fiber,RRF),光镜下RRF在Gomori染色中红染、SDH染色中深染,可见呼吸链复合物(COX)阴性纤维,
表明细胞色素氧化酶活性缺失,不同COX活性的纤维间隔排列,呈“马赛克”样。乳酸升高较少见。
发病年龄
CPEO为线粒体大片段缺失突变中较为常见的表型,任何年龄均可发病,诊断年龄为0岁以上的任何年龄阶段,
以 41岁以上居多,其次为 20~40 岁,20岁以下较少,尤其是5岁以下。
KSS
临床表现
KSS是一种多系统受累的 20岁以前发病的线粒体脑肌病,临床表现以进行性眼外肌麻痹和视网膜色素变性为主,
如果伴心脏传导阻滞、共济失调和脑脊液蛋白增高 3 项中任何 1项时称为 KSS三联征。
除了进行性眼外肌麻痹的表现,色素性视网膜病变是KSS的重要临床特点,典型眼底改变为“椒盐状”(salt and pepper),即视网膜广泛色素改变,
包括颗粒状色素沉着和脱色素,症状最初比较轻微,但随着年龄增长会变得愈发典型而广泛,少数可进展到骨细胞样色素改变,此外,还有脉络膜毛细血管萎缩表现。
“椒盐状”视网膜病变的患者早期视功能通常不受影响,无明显视力下降,其眼底检查异常早于视网膜电图和眼电图出现。视网膜电图显示视网膜营养不良,视野检测无异常。
鉴别诊断
KSS 被认为是与 CPEO 有共同遗传基础的线粒体脑-肌病,是Kearn和Sayre在1958年首次报道的1种散发的多系统受累的线粒体脑-肌病。
临床根据20岁以前起病、进行性眼外肌麻痹和色素性视网膜病变,并至少出现以下症状之一:心脏传导阻滞、小脑共济失调、脑脊液蛋白增高(>100 mg/L)即可临床诊断。
KSS与 CPEO 同属 MD的同一种综合征的不同表现,当 CPEO发生在 20岁以前,伴视网膜色素变性、脑脊液蛋白增高、共济失调或心脏传导阻滞时称为 KSS ,
KSS是一种更严重的、变异的 CPEO 。
KSS临床异质性高,几乎都存在眼部症状,但不一定以眼部症状起病,且不一定在就诊时已经出现视网膜色素变性,需定期随访。
有些CPEO 的病例中也会出现色素性视网膜病,但较KSS轻微。
分子诊断
致病机制
KSS和CPEO是由于mtDNA缺失或重复突变致病,多为mtDNA的2~10 kb缺失,90%的KSS在所有组织中存在大片段(1.1~10.0 kb)的 mtDNA 缺失,已知与之相关的超过 150 种缺失或重复突变,
最常见的为 mtDNA 8470~13446 的4977bp 缺失。此外,部分 KSS 患者还存在大片段重复。
检测手段
既往认为,KSS 和 CPEO 的分子诊断主要依赖于肌肉组织活检,病理发现 RRF 以及在肌肉组织中发现 mtDNA 缺失或重复突变而确诊,在外周血和尿液标本中通常不能检测到突变。
曾有报道 KSS 或 CPEO 只能在肌肉组织中检测到 mtDNA的大片段缺失或重复。可能与当时检测手段敏感度过低或外周血细胞中不存在突变(或突变比例过低)有关。
近年来,随着二代测序技术的出现和不断发展,通过外周血或尿液检测大片段缺失来进行诊断 KSS 成为可能,对于没有发现基因突变的,依然需要肌肉活检。
KSS 和 CPEO 一般为散发病例,少见有家族史。
外显率
在mtDNA相关疾病中,外显率是异常mtDNA分子比例的函数。一般情况下,当异质水平超过80%-90%时,可引起线粒体功能障碍和临床症状。
然而,现在人们认识到,这是高度可变的,取决于突变的位置和严重程度、检测的特定的组织、进行检测的年龄和使用的检测方法。
线粒体疾病受mtDNA和nDNA双重调控而致病的遗传代谢性疾病。
由于临床表现多样,缺乏特定的生物学标记物,涉及2个基因组的遗传方式,给基因诊断带来困难。
韦翰斯生物检测方案
备注:临床怀疑是mtDNA片段缺失导致的线粒体缺失综合征,建议送检受检者的组织,结合long PCR+NGS做线粒体高深度测序。



今年一月,一对年轻的夫妇在初步知悉了蕴可安™无创产前检测项目后联系了韦翰斯,在沟通后我们得知:他们已经育有一名患有耳聋的孩子,现在妻子再次怀孕,非常担心自己即将出生的孩子罹患相同的疾病。摊开一沓厚厚的诊疗资料,我们了解到这个耳聋的孩子患有双耳极重度感音神经性聋。正常个体能够听到的最小声音范围在0-25分贝之间,而极重度听力损失的患者只能听到90分贝以上的声音;要知道,90分贝相当于闹市区车水马龙的大街上的响度,而这仅仅是患者感知声音的开始。
鉴于这个家庭之前从未进行过基因检测,我们先建议他们以孩子为先证者进行家系临床全外显子组测序项目查明病因,结果发现孩子身上携带了TMC1基因上的2个杂合变异。经过一代测序验证后证实了分析结果的可靠性;这两个变异分别来自其父亲和母亲,符合受检者无家族史的描述。

先证者的临床全外显子组测序结果
古语有曰:“往者不可谏,来者犹可追。”当下这对年轻的夫妇能做的就是避免自己的二胎也降临到一个无声的世界上。以往常见的孕中胎儿基因检测需要通过羊水穿刺手段取样,羊水穿刺不但会为孕妇带来流产的风险,同时还可能引起早产、感染、母体免疫反应等并发症;更何况本案例中的准妈妈孕期较短,没有达到16-23周的羊水穿刺实施标准——相较之下,韦翰斯独有的蕴可安™无创产前单基因病检测项目不但更安全、更可靠,还能早在怀孕8周就通过无创的手段检测胎儿患病的风险。
基于对韦翰斯专业性的信任,这对夫妇选择了蕴可安™无创产前检测;就像常规基因检测那样,我们仅仅取用孕妇及核心家系的外周血,经过认真、严谨的实验室分析流程及检测结果解读之后,于15个工作日之内出具了详实可靠的检测报告——萦绕在受检夫妇心头的疑问最终随着阴性结果的到来而烟消云散:未在胎儿身上发现携带母源或父源的致病变异。
检测结果表:

胎儿的无创产前检测结果

韦翰斯是一家活跃在基因检测第一线的公司,旨在坚持不懈地提供各类优质的遗传病检测服务,为更多的家庭带去健康的孩子。
蕴可安™无创产前单基因病检测项目覆盖了2000多个基因相关的、致病机制明确的、2500多种单基因遗传病;可以针对任何有先证者的家系(先证者父母双方或一方为相同致病基因变异的携带者),在极短周期内做到准确高效的检测;除此之外,该项目在孕早期(怀孕8周)即可检测,为产前诊断留出了充足的时间,同时检测准确性>99%,远高于目前其他常规产前筛查项目;本项目采用外周血作为检测样本,避免羊水穿刺活检、绒毛膜活检和脐带穿刺术等传统取样方法带来的一系列安全隐患,真正做到又快、又准、又安全。
无创产前单基因病检测技术不同于传统的NIPT,主要是通过采取孕妇外周血,利用二代DNA测序技术对母体外周血中的游离DNA片段(cell-free DNA,cfDNA)进行测序,并将测序结果进行数据化处理、生物信息分析,最终解读出胎儿是否遗传父母的单基因突变,对降低出生缺陷发生起到非常重要的作用。目前,单基因病的无创产前检测逐渐成为全球NIPT临床应用的发展趋势。
早发性高度近视是一种发生在学龄前儿童中的眼科疾病,基因遗传是主要的致病原因。

根据截止2021年的统计,人类孟德尔遗传数据库(OMIM)在线收录的相关基因有20个,基因突变引发的眼科疾病涵盖了多种遗传模式,不同疾病间的症状又常有重叠;因此基因检测和遗传分析对患者的早期诊断、精准治疗及预后处理至关重要,同时也对进行遗传分析与咨询的专业人士提出了巨大的挑战。
此外,随着变异筛选和评级软件的不断推陈出新,自动化的遗传学分析已经占据了越来越高的比重;在享受其带来的经济、便捷的同时,也出现了漏检一些极为罕见病症的可能性,因此人工进行突变分析有其不可替代的必要性。

今天我们带来了一篇针对早发性高度近视患者进行遗传检测的案例分享:
女孩在2岁时就已经出现了近视表型,3岁时在其他单位进行了第一次基因检测,结果显示患者及其父亲PRPF6基因位点上发生了相同的缺失-插入突变,导致了常染色体显性遗传的视网膜色素变性60型。在后续的复诊过程中,经验丰富的坐诊教授发现患者表现出来的症状与视网膜色素变性多有出入:不但电生理指标不吻合,而且患者也不具有夜盲症的表型。

出于对患者负责到底的理念,医生和患者父母沟通后一致决定再次进行基因检测,这一次他们选择了韦翰斯。我们在收到细心存运的受检者样本后,实验室按照严格的检测流程分析出了有关患者基因的原始数据,随后专业的遗传咨询团队一丝不苟地整理并寻找着有价值的信息。
事实果然如同教授预料的那样,患者的症状背后另有隐情:我们成功在患者的ARR3基因上找到了一处关键的杂合无义变异位点,可能导致基因功能缺失,并通过一代测序验证显示受检者父母均未检出该变异;虽然从严谨的角度出发,仅根据ACMG指南的评级结果不足以直接为该突变的致病性盖棺定论,但是新发变异的确认大大增加了该突变致病的可能性,使其成为一条重要的线索、一个一探究竟的机会。
位于染色体Xq13.1上的ARR3基因异常会导致X连锁女性限制性近视26型,这是一种非常罕见的眼科疾病,在OMIM数据库中仅有一篇研究了3个中国家系的文献报道,该病仅限女性发病:在三个家族中,所有杂合子女性成员都受到影响,而所有半合子男性家庭成员都不受影响(Xiao等人,2016),并且国内外对该病的致病机理也尚不明确;能够探明如此罕见的致病原因并合理解释患者出现的表型,离不开我们的遗传咨询团队始终坚持对突变位点致病性进行人工评级的努力,越是既往报道发现少见的情况越是难以通过自动化的分析手段实现精准判断,我们的坚守再一次证明了:功不唐捐。

新位点的发现不仅拨开了患者及家属心头萦绕的迷雾,同时还为临床研究标记出了一条新的研究思路:对本次发现的突变位点的功能性研究或许可以揭示ARR3基因和对应疾病之间的关联,进一步明晰致病机制。早发性近视本身对于患儿来说就是成长路上的一条拦路虎,更何况本案例中的女孩于两岁就罹患眼疾,处于大脑高速发展期的孩子睁眼望见的却是一片模糊不清、含混迷离的世界,是何等的不幸!假如在患儿第一次检测中便能明晰病因,又能够在病理更为明朗、预后更加精确的基础上及早干预,也许人生会大有不同。
本次基因检测结果除了发现与受检者临床表型相关的基因变异之外,更进一步地发现了DSC2基因上可能致病的1个杂合移码变异,DSC2基因异常可导致常染色体显性/隐性遗传的致心律失常性右室发育不良11型。这种心肌疾病在4-64岁间均可发病,以往病例的平均诊断年龄高达31岁,其隐匿性可见一斑;与此同时这种心肌病又容易导致年轻个体发生室性心动过速和猝死,足以说明起病迅猛;联系起这两个特点就好像能看见病人头顶上的一柄悬剑,不知何时会陡然落下。好在当我们提前知晓病人面临着心肌病的风险时,就可以及时向这个家庭提供具体的临床建议,包括定期对心脏功能和心搏节律进行检查、劝阻剧烈活动并给出日常预防的种种手段等,哪怕有一天发现了病情进展的征象也可以结合药物管理和手术治疗来预后,尽早发现潜在的风险是对患者及其家属生理和心理上的双重保障。

来自临床医生对韦翰斯的肯定

本次用于检测的项目为全外显子组测序(含线粒体),简称为WES;WES项目包含了对人类20000多个基因的全部外显子检测,平均的测序深度大于100×,其中30×以上覆盖了超过98%的区域,还有对线粒体全长的测序,平均深度大于1000×;主要分析的变异类型为点突变、小的插入缺失、杂合性丢失以及大于3个连续外显子的拷贝数变异 。覆盖全部基因编码区域的筛查布下了天罗地网,不仅让临床怀疑的重点检测对象无所遁形,还能根据ACMG的官方建议对一些发病较晚或预后较好的其他疾病做出风险提示(次要发现)以及报告与患者表型相关的一些突变位点(附录-其他变异),并结合一代测序对患者父母的相关位点进行验证,以明确患者突变的来源,为临床提供更加多元的、详尽的诊断线索。

在接下来的过程里我们会继续坚持严谨的人工筛选及评级标准,以最专业负责的态度做好基因检测的每一步,用优质的服务品质来落实“让每一个家庭都拥有健康的孩子”之理念。

值得一提的是,对于本例患者这种临床表型复杂难以确诊的情况,比上述的“先证者模式”更为契合的是“家系模式”:家系模式检测会直接对患者及其父母的基因进行全面的筛查,如果把单凭先证者测序结果和父母的一代验证进行的分析推测比作为“顺藤摸瓜”的话;拥有全部三人测序结果后便是“开门见山、一目了然”,对于患者的突变类型和致病性的读判不再依赖略显模糊的表型,而是根据父母相应位点的突变情况进行严格的遗传学解释,这样一来不仅结果直观确凿还大幅提升了置信度,可谓是真正意义上的又快又准。

文章名:一例伴发多脏器畸形的新生儿糖尿病患者的临床及基因变异分析
DOI: 10.3760/cma.j.cn511374-20190814-00409
期刊:中华医学遗传学杂志, 2020,37(12) : 1371-1375.
目的:分析1例伴发多脏器畸形的新生儿糖尿病患儿的基因变异特点,明确其致病原因。
方法:应用全外显子组测序对患儿进行分析,并用Sanger测序进行验证。
结果:测序结果显示患儿GATA6基因第5外显子存在c.1454_1455del(p.K485Rfs)杂合变异,既往未见文献报道,患儿父母均未发现相同变异。
结论:GATA6基因c.1454_1455del(p.K485Rfs)杂合变异可能为患儿的致病原因,上述结果为该家系的遗传咨询和产前诊断提供了依据。
文章截图:

文章名:克氏综合征合并X染色体隐性遗传家族性眼震一例
期刊:中华医学遗传学杂志2021年11月第38卷第11期
DOI:10.3760/cma.j.cn511374-20201102-00767
文章截图:

文章名:Epimutation
of MMACHC compound to a genetic mutation in cblC cases
参考链接:https://doi.org/10.1002/mgg3.1625
网站/文章截图:

文章名:Analysis of NGS-Ministr as A NewDetection StrategyChromosomal Trisomy 16 of Fetal Materials in Miscarriages
DOI:https://www.researchsquare.com/article/rs-658285/v1
杨敬敏博士:韦翰斯生物联合创始人CTO
网站/文章截图:

文章名:Characterization of Unique LensMorphology in a Cohort of Children with Familial Exudative Vitreoretinopathy
期刊:Current Eye Research


文章名:Whole-exome sequencing identifies a
novel mutation in spermine synthase gene (SMS) associated with Snyder-Robinson
Syndrome
期刊:BMC Medical Genetics
网站/文章截图:


文章名:Establishment of non-integrate inducedpluripotent stem cell line CSUASOi006-A, from urine-derived cells of aPRPF8-related dominant retinitis pigmentosa patient
参考网址:https://www.sciencedirect.com/science/article/pii/S1873506120303421
网站/文章截图:


这样的对话每天都在发生,韦翰斯遗传病基因检测是认真的,我们要再接再励。
文章名:25例先天性高胰岛素血症临床特征及随访分析
发布在中国小儿急救医学 2019 年 7 月第 26 卷第 7 期 Chin Pediatr Emerg MedJul 2019Vol. 26No. 7
参考网址:http://rs.yiigle.com/CN115454201907/1155180.htm
文章截屏:

文章名:SDHB基因新发剪接突变导致下腔静脉旁副神经节瘤一例分析
发布在中华内分泌代谢杂志, 2020,36(02) : 153-155. DOI: 10.3760/cma.j.issn.1000-6699.2020.02.012
参考网址:http://rs.yiigle.com/CN311282202002/1182455.htm
网站截屏:

文章名:A Novel Mutation in the NDP Gene is Associated with Familial Exudative Vitreoretinopathy in a Southern Chinese Family
参考网址:https://www.liebertpub.com/doi/10.1089/gtmb.2019.0099
网站截屏:
参考网址:https://www.sciencedirect.com/science/article/pii/S2352187219300464
网站截图

受检者临床基本信息:
患者由于继发性闭经为主诉,妹妹有原发性闭经
检测方案:
韦翰斯遗传病检测项目
检测结果:
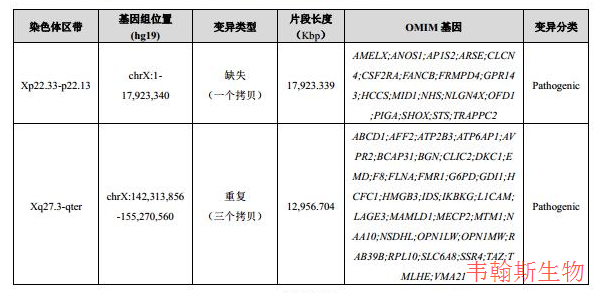
检测分析,发现X染色体上存在两个与患者临床表型相关的拷贝数变异,M1为X染色体短臂远端缺失(Xp22.33-p22.13),该缺失可能导致Turner综合征。Turner综合征(Turnersyndrome, TS,又称特纳综合征)是导致女孩身材矮小及年轻女性原发性闭经的一个重要病因,通常由一条X染色体的部分或全部缺失所引起。Turner综合征在活产女婴中的发病率约为 1/2500~1/3000。Xp22缺失与身材矮小、点状软骨发育不良、鱼鳞病、智力缺陷及Kallmann综合征等有关。
M2导致Xq27.3-q28重复综合征,该综合征主要临床表现有:身材矮小,腹部肥胖,小手小脚,轻度精神发育迟滞,原发性性腺功能减退,男性乳房发育症、小阴茎、隐睾,女性携带者身材矮小、卵巢早衰,高促性腺激素、低睾酮等。
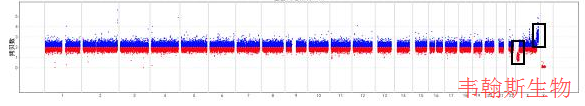
为进一步确认,要求受检者及父母进行染色体核型分析、FISH(荧光原位杂交技术)等技术检测,临床需根据患者表现和家族史做进一步判断。
受检者临床基本信息:
患者临床表现为低钾血症、碱血症。临床怀疑Gitelman综合征或Batter综合征。
检测方案:
韦翰斯遗传病检测项目
检测结果:
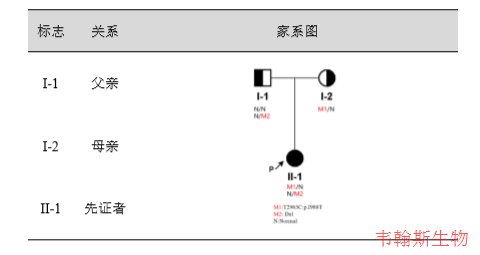
发现先证者 SLC12A3 基因的一个杂合变异c.T2963C:p.I988T,对亲属的一代测序结果显示,先证者父亲未携带该变异,先证者母亲为杂合变异,推测该变异遗传自母亲,结果如下所示:

对二代测序数据进行拷贝数变异分析,提示先证者16号染色体SLC12A3基因可能存在约10.43Kbp的杂合缺失,该缺失区域包含SLC12A3基因第20-24号外显子。对一家三口该区域进行qPCR验证,发现先证者和其父亲为杂合缺失,母亲无缺失。推测该拷贝数变异遗传自父亲,结果如下所示:
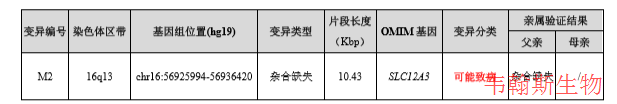
SLC12A3 基因异常可导致常染色体隐性遗传的Gitelman综合征(Gitelman Syndrome; GTLMNS,OMIM#263800)
变异验证:
一代测序对先证者及亲属SLC12A3基因变异M1位点进行验证,结果显示该变异真实可靠, 先证者父亲未携带该变异,先证者母亲为杂合变异。
qPCR 对先证者及亲属 SLC12A3基因的缺失区域进行验证:在SLC12A3基因缺失区域设计三个扩增子,缺失区域的上下游各设计一个对照扩增子,X染色体上设计一个对照扩增子,使用qPCR对以上扩增子进行相对DNA定量。qPCR结果显示先证者及其父亲为杂合缺失。
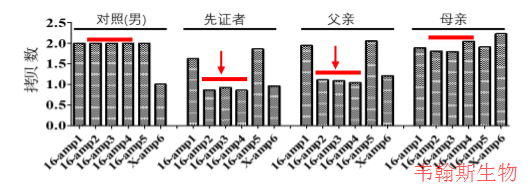
遗传咨询:
Gitelman 综合征(Gitelman syndrome, GS)是Bartter综合征的一种变异型,又称伴低尿钙、低血镁的Bartter综合征,属常染色体隐性遗传的肾小管疾病。对于患儿而言,最常见的症状包括嗜盐,夜尿增多及明显的低钾低镁血症相关的肌肉乏力和抽搐发作。通常认为Gitelman综合征的临床表现很轻,有些患者甚至终生无明显症状,可对症治疗。对于夫妻双方,明确自身突变的携带情况,再次生育时应做好产前诊断或者是胚胎植入前检测,避免再次生育患儿。